Montag bis Freitag von 08:00 bis 15:00 Uhr

Online-Dienst
Medizinprodukte in Europa online
Gesamtpaket der harmonisierten und mandatierten Normen
Stand 2024-04
- Autoren
- Dipl.-Ing. (FH) Elisabeth Beck, Dr. Rainer Edelhäuser
- Information
-
Dieses Produkt bietet DIN Media ausschließlich Kund*innen an, die keine Verbraucher im Sinne des § 13 BGB sind.
Nutzung nur im Abonnement möglich. Die Bestellung bewirkt ein Abonnement für mindestens ein Jahr. - Der Vertrag verlängert sich jeweils um ein weiteres Jahr, wenn er nicht mit einer Frist von 3 Monaten zum Ende des jeweiligen Verlängerungszeitraums gekündigt wird.
- Hinweis
- Dieses Produkt enthält mind. eine Norm mit Warnvermerk. Die betroffene Norm finden Sie in dieser Liste .
- Autoren
- Dipl.-Ing. (FH) Elisabeth Beck, Dr. Rainer Edelhäuser
- Information
-
Dieses Produkt bietet DIN Media ausschließlich Kund*innen an, die keine Verbraucher im Sinne des § 13 BGB sind.
Nutzung nur im Abonnement möglich. Die Bestellung bewirkt ein Abonnement für mindestens ein Jahr. - Der Vertrag verlängert sich jeweils um ein weiteres Jahr, wenn er nicht mit einer Frist von 3 Monaten zum Ende des jeweiligen Verlängerungszeitraums gekündigt wird.
- Hinweis
- Dieses Produkt enthält mind. eine Norm mit Warnvermerk. Die betroffene Norm finden Sie in dieser Liste .
Produktinformationen auf dieser Seite:

Medizinprodukte in Europa online versammelt die wichtigsten Normen und Richtlinien zum Thema, auf die Sie online immer und von überall zugreifen können. Durch den stetigen Austausch mit Expert*innen wird sichergestellt, dass die Inhalte die jeweiligen Branchenanforderungen widerspiegeln.
Enthalten sind unter anderem die rechtlichen Grundlagen für das europaeinheitliche technische Regelwerk zu Medizinprodukten, die Verordnungen der Europäischen Union (MDR, IVDR) und eine Sammlung der wichtigsten Medizinprodukte-Normen.
Vierteljährliche Updates (ca. Februar, Mai, August, November) bringen alle Inhalte regelmäßig auf den aktuellen Stand. Zum aktuellen Update-Verzeichnis.
Ihre Vorteile:
- Normen und Rechtsvorschriften einfach auf dem PC oder Tablet aufrufen
- Vierteljährliche Updates inklusive – keine Änderung verpassen
- Großer Preisvorteil gegenüber dem Kauf von Einzelnormen
- Separate Themenmodule erhältlich – Sie entscheiden, was Sie brauchen
- Rechtskonforme Nutzung von Normen durch optionale Mehrplatzlizenzen
- Auch als Pro-Version mit nützlichen Funktionen: Speichern, Drucken, Filtern
- Historischer Pool mit zurückgezogenen Normen
Zielgruppe
Herstellende Unternehmen von Medizinprodukten, Prüfstellen, Aufsichtsbehörden und Anwendende von Medizinprodukten.
So arbeiten Sie mit dem Online-Dienst in der Pro-Version:

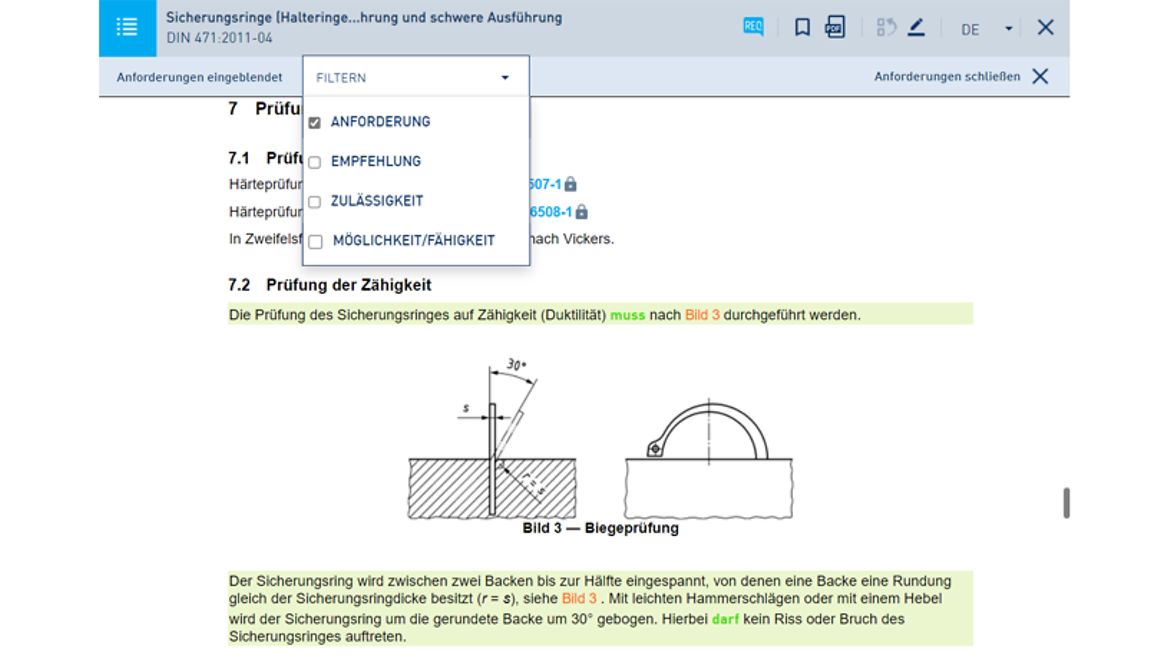
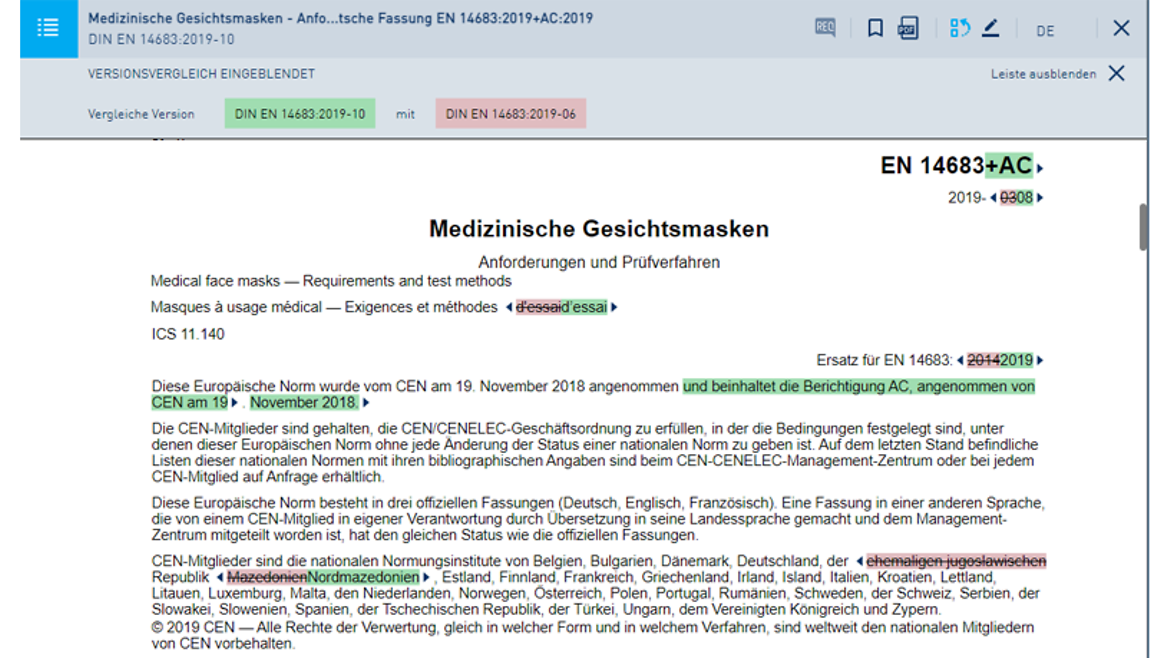
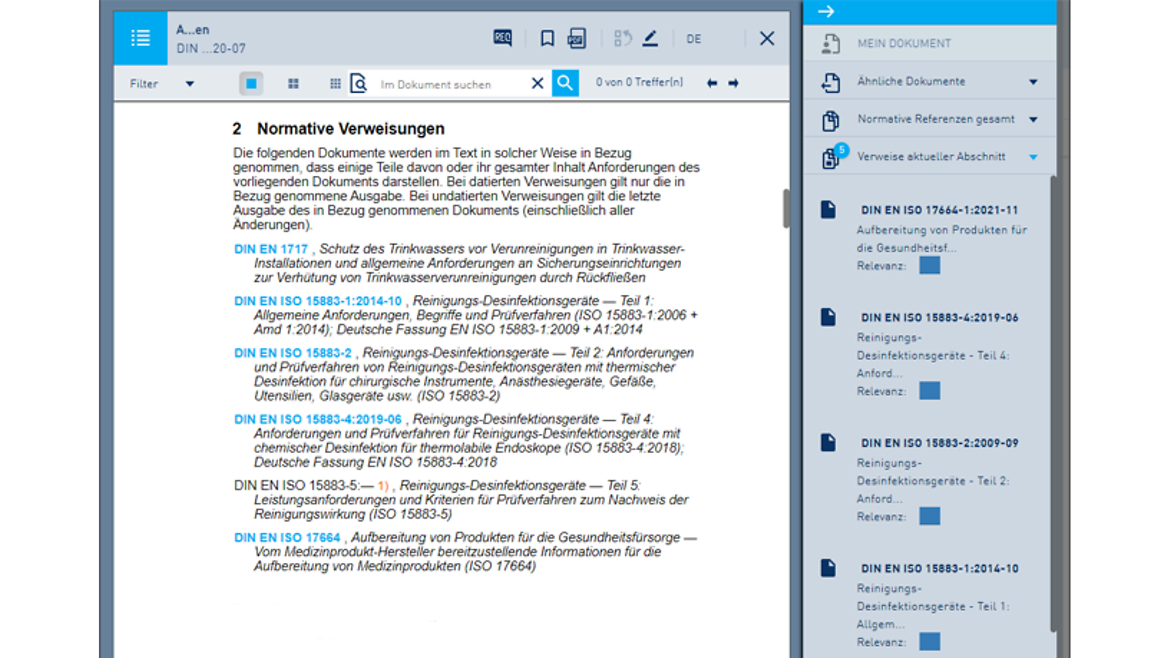
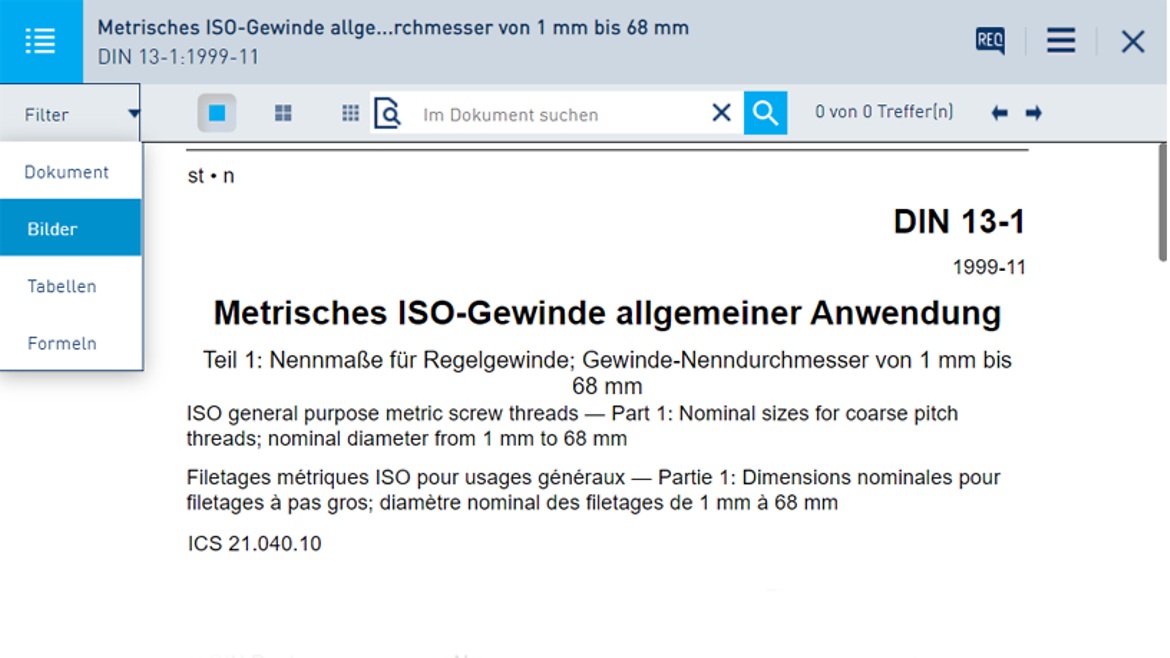
Eine Übersicht über alle Online-Dienst-Funktionen finden Sie hier.
Rabattaktion auf den Abo-Jahrespreis
Sie haben noch kein Abo?
Dann nutzen Sie jetzt unsere Rabattaktion und erhalten Sie 20 % Preisnachlass im ersten Bezugsjahr.
Der Rabatt wird nicht im Buchungsprozess angezeigt, sondern auf der Abrechnung ausgewiesen. Sie können ihn nur nutzen, wenn Sie in den vergangenen 24 Monaten weder diesen noch einen anderen Online-Dienst des Beuth Verlags abonniert hatten.
Sie haben bereits ein Abo?
Dann steigen Sie jetzt auf die Pro-Version oder auf eine Mehrplatz- oder Standortlizenz um und erhalten Sie 20 % Rabatt im ersten Bezugsjahr. Nutzen Sie viele praktische Funktionen, zum Beispiel das Speichern und Ausdrucken von Dokumenten.
Wenn Sie Ihrem Abo ein weiteres Modul hinzufügen möchten, erhalten Sie darauf ebenfalls 20 % Rabatt.
Der Rabatt wird nicht im Buchungsprozess angezeigt, sondern auf der Abrechnung ausgewiesen.
Noch unentschlossen?
Lernen Sie in 30 Minuten, wie Sie mit dem Normen-Abo effizienter arbeiten. In unserem kostenlosen Webinar stellen wir Ihnen alle Funktionen vor und beantworten Ihre Fragen.
Das Gesamtpaket der harmonisierten und mandatierten Normen enthält die in Europa zu beachtenden wesentlichen Rechtsbestimmungen und harmonisierten und mandatierten Normen für Medizinprodukte (inkl. die aller Themenpakete).
ab 61,58 EUR/Monat inkl. MwSt.
ab 739,00 EUR/Jahr inkl. MwSt.
Das Themenpaket enthält sowohl die harmonisierten und mandatierten als auch nicht mandatierte bzw. harmonisierte Normen zu folgenden Themengebieten: Sterilisation von Medizinprodukten, Reinigungs- und Desinfektionsgeräte, Sterilisatoren für medizinische Zwecke und Verpackungen für in der Endverpackung zu sterilisierende Medizinprodukte.
ab 34,67 EUR/Monat inkl. MwSt.
ab 416,00 EUR/Jahr inkl. MwSt.
Das Themenpaket Beatmungs- und Anästhesiegeräte enthält alle mandatierten und harmonisierten Normen zu diesem Bereich einschließlich Geräteanforderungen, Sicherheit und wesentliche Leistungsmerkmale oder Prüfverfahren.
ab 31,75 EUR/Monat inkl. MwSt.
ab 381,00 EUR/Jahr inkl. MwSt.
Das Themenpaket Biokompatibilität von Medizinprodukten enthält die für die Beurteilung der Biokompatibilität von Medizinprodukten grundlegende Reihe DIN EN ISO 10993. Alle Normen dieser Reihe sind harmonisiert oder mandatiert.
ab 20,75 EUR/Monat inkl. MwSt.
ab 249,00 EUR/Jahr inkl. MwSt.
Das Themenpaket Qualitätsmanagement und allgemeine Aspekte für Medizinprodukte enthält die grundlegenden Normen für alle Anwender, die mit Medizinprodukten zu tun haben: Hersteller, Zulieferer, Beratungsunternehmen, Benannte Stellen etc.
Außerdem sind in diesem Paket die Europäischen Verordnungen und Richtlinien mit enthalten, die sonst nur Teil des Gesamtpakets der harmonisierten und mandatierten Normen mit sind.
ab 15,92 EUR/Monat inkl. MwSt.
ab 191,00 EUR/Jahr inkl. MwSt.
Das Themenpaket In-vitro-Diagnostika enthält alle harmonisierten oder mandatierten Normen zu In-vitro-Diagnostika und wendet sich somit gezielt an Hersteller und Prüfungsinstitute von In-vitro-Diagnostika.
ab 18,92 EUR/Monat inkl. MwSt.
ab 227,00 EUR/Jahr inkl. MwSt.
Das Themenpaket Nichtaktive chirurgische Implantate enthält mandatierte und harmonisierte Normen unter anderem aus den Bereichen allgemeine Anforderungen, kardiovaskuläre Implantate und Endoprothetic.
ab 22,83 EUR/Monat inkl. MwSt.
ab 274,00 EUR/Jahr inkl. MwSt.
Das Themenpaket Nichtaktive Medizinprodukte enthält mandatierte und harmonisierte Normen unter anderem aus den Bereichen Infusionsgeräte für medizinische Verwendung, Injektionsgeräte und Spritzen sowie Katheter.
ab 34,67 EUR/Monat inkl. MwSt.
ab 416,00 EUR/Jahr inkl. MwSt.